


When a patient with rheumatoid arthritis or cancer needs a biologic drug, they’re often faced with a price tag of $50,000 to $100,000 a year. That’s not a typo. These drugs save lives, but they’re also among the most expensive medicines in the U.S. healthcare system. Enter biosimilars-medicines that are not exact copies, but are so close in structure and function that they work the same way in the body. And since October 2025, the FDA has made it significantly easier for these lower-cost alternatives to reach patients.
What Makes a Biosimilar Different From a Generic Drug?
Think of generics as the straightforward twins of brand-name pills. If you take a generic ibuprofen, it’s chemically identical to Advil. The molecule is the same. The manufacturing process is simple. The FDA can approve it by comparing chemical structure and bioavailability.
Biosimilars are not like that. They’re made from living cells-yeast, bacteria, or mammalian cells grown in labs. These cells produce complex proteins like antibodies or hormones. Even tiny changes in temperature, pH, or nutrient mix during production can alter the final molecule. That’s why you can’t just copy a biologic like you copy a tablet. A biosimilar must be highly similar, not identical. And proving that similarity takes advanced science.
The FDA requires data from analytical tests, pharmacokinetic (PK) studies, and sometimes clinical trials. But here’s the big shift: as of October 2025, the FDA no longer automatically requires full comparative efficacy studies. That’s a game-changer.
The FDA’s 2025 Guidance: A New Path to Approval
Before October 2025, getting a biosimilar approved often meant running a three-year clinical trial comparing it head-to-head with the original biologic. That cost between $100 million and $300 million. Only a handful of big companies could afford it.
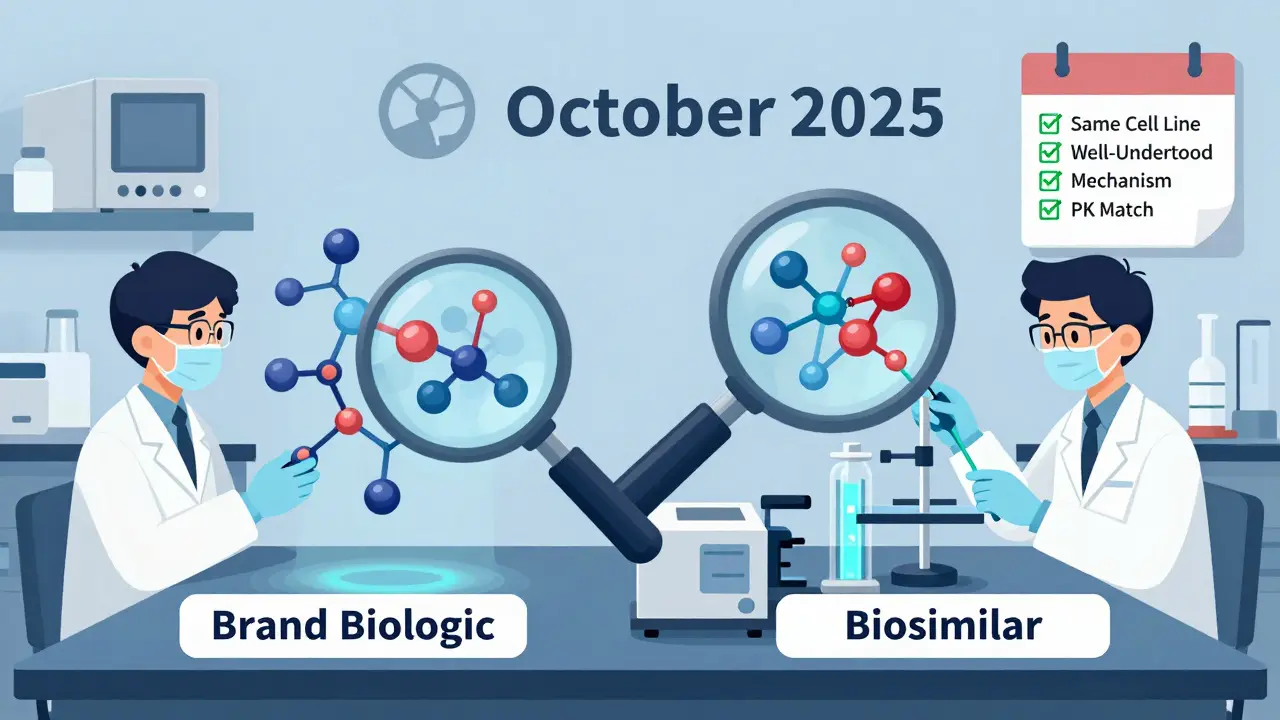
The new draft guidance from the FDA changed that. Now, if three conditions are met, you might not need a large efficacy trial at all:
- The reference biologic and the biosimilar are made from the same type of cell line and are highly purified.
- The link between the molecule’s structure and how it works in the body is well understood-for example, with monoclonal antibodies like adalimumab or trastuzumab.
- You can run a reliable pharmacokinetic study showing the drug behaves the same way in the bloodstream.
If those boxes are checked, the FDA says analytical data from mass spectrometry, chromatography, and bioassays can be enough. These tools can now detect differences smaller than one part per million. That’s more precise than any clinical trial measuring patient outcomes over months.
This isn’t just theory. The FDA approved two denosumab biosimilars with interchangeability status in October 2025-the first time multiple interchangeable biosimilars were approved for the same reference product. That’s a milestone.
Interchangeability: What It Means and Why It’s Controversial
Interchangeability is the holy grail for biosimilars. It means a pharmacist can swap the biosimilar for the brand-name drug without asking the doctor. In 34 states, that’s still not allowed-even if the FDA says it’s safe.
The FDA’s new stance? Commissioner Marty Makary said at a conference in October 2025: “Every biosimilar should have the designation of interchangeable.” He called interchangeability a “legislative term, not a scientific term.” That’s bold. It suggests the current legal requirement for separate switching studies is outdated.
But not everyone agrees. Some doctors and pharmaceutical industry leaders worry removing the interchangeability hurdle could erode trust. A study from the Arthritis Foundation found 41% of patients were initially scared to switch. Only after talking to their doctor did most feel comfortable.
Meanwhile, the FDA continues to grant interchangeability status on a case-by-case basis. The October 2025 approvals for denosumab biosimilars prove the system still works-but the push to make it automatic is creating tension between science and law.
How the FDA Reviews Biosimilars Today
The process starts with a Biologics License Application (BLA) under Section 351(k) of the Public Health Service Act. Here’s what the FDA looks for:
- Analytical characterization: Over 200 quality attributes are measured-protein folding, sugar chains, impurity profiles. Tools like liquid chromatography-mass spectrometry (LC-MS) are standard.
- Non-clinical studies: Toxicity and immunogenicity tests in animals to check for unexpected immune reactions.
- Pharmacokinetic studies: Blood samples taken over time to compare how fast and how long the drug stays in the body.
- Clinical studies: Now only required if the above data isn’t sufficient. For most monoclonal antibodies, they’re often skipped.
It’s not easy. About 42% of biosimilar applications get a “complete response letter” from the FDA, meaning more data is needed. But with the new guidance, that number is expected to drop.
Who’s Making Biosimilars-and Who’s Not
As of late 2025, the FDA has approved 76 biosimilars. But the market is dominated by just a few players:
- Sandoz: 17 approved biosimilars
- Pfizer: 12
- Amgen: 10
Smaller companies struggle. Only 12 of the 76 approved biosimilars came from firms with fewer than 100 employees. Why? Because setting up the analytical labs needed for biosimilar development costs tens of millions. A small biotech can’t afford the equipment, the staff, or the time.
That’s changing slowly. The FDA’s Biosimilars Community Resource Center had over 12,000 visitors in October 2025. The Biosimilars Council has offered 87 free consultations to small developers since 2023. And the BsUFA III funding program, which runs through 2027, is helping speed up reviews.

Market Reality: Why Biosimilars Still Aren’t Everywhere
Even with FDA reforms, biosimilars make up only 23% of the market for drugs with alternatives. In Europe, that number is 67%.
Why the gap?
- Patent litigation: The FTC says 68% of approved biosimilars face legal delays. Big pharma uses patents to block entry-even for minor changes.
- Physician hesitation: Many doctors still prefer the original drug, even if it costs more.
- Patient awareness: Only 32% of patients know what a biosimilar is, according to the National Biosimilars Survey.
- State laws: 34 states still require prescriber approval before substitution, even when the FDA says it’s safe.
But change is happening. Mayo Clinic saved $18 million in a year by switching oncology patients to biosimilars. Hospital formularies now include biosimilars in 89% of U.S. hospitals. And the market is projected to grow from $18.7 billion in 2024 to $62.3 billion by 2029.
What’s Next for Biosimilars?
The FDA’s draft guidance is open for public comment until January 27, 2026. Final rules are expected by June 2026. Analysts predict annual approvals could jump from 8-10 to 15-20 per year.
But two big hurdles remain:
- Legislative action: If the FDA wants to make all biosimilars interchangeable by default, Congress may need to change the law. Right now, the law requires separate approval for interchangeability.
- Complex molecules: Antibody-drug conjugates and other advanced biologics are harder to characterize. The new guidance doesn’t fully solve that yet.
Still, the direction is clear. The FDA is moving from a trial-heavy model to one based on science, not bureaucracy. And that’s good news for patients who need life-saving drugs but can’t afford the price tag.
Real Patient Experiences
On Reddit’s r/pharmacy, over 80 people shared their experiences switching to biosimilars for autoimmune diseases. Sixty-three percent said their symptoms stayed the same. Twenty-two percent noticed minor differences-mostly injection site reactions. No one reported serious safety issues.
One patient wrote: “I switched from Humira to the biosimilar. Same results. Paid $20 instead of $3,000. My insurance didn’t even notice.”
That’s the goal: safety, effectiveness, and affordability-all in one.
Are biosimilars as safe as the original biologic drugs?
Yes. The FDA requires extensive testing to prove biosimilars have no clinically meaningful differences in safety, purity, or potency. Since 2015, over 76 biosimilars have been approved in the U.S., and post-market surveillance has not shown any new safety risks compared to the original biologics. Real-world data from hospitals and patient surveys confirm similar outcomes.
Can pharmacists automatically substitute a biosimilar for a brand-name drug?
Only if the biosimilar has an FDA interchangeability designation AND your state allows automatic substitution. As of 2026, 16 states permit pharmacists to switch without prescriber approval. The other 34 still require a doctor’s permission, even if the FDA says it’s safe. This mismatch between federal and state rules creates confusion for patients and pharmacies.
Why are biosimilars cheaper than biologics?
Biosimilars cost less because they don’t need to repeat the original clinical trials that proved the biologic works. The FDA’s updated 2025 guidance allows manufacturers to rely on analytical data and smaller studies, cutting development costs from $100-300 million to $50-150 million. That savings gets passed on-biosimilars typically cost 15-35% less than the reference product.
Do biosimilars work for all conditions that biologics treat?
Biosimilars are approved for the same uses as their reference biologics-like rheumatoid arthritis, Crohn’s disease, diabetes, and certain cancers. But they’re not approved for every condition the original drug treats unless the manufacturer proves similarity for each use. Most biosimilars are approved for all indications of the reference product, but not always. Always check the FDA-approved labeling.
How long does it take to get a biosimilar approved now?
Before 2025, the process took 8-10 years. With the new FDA guidance, it’s now 5-7 years on average. The biggest time-saver is skipping full comparative efficacy trials. The FDA’s BsUFA III program also ensures reviews are completed within 10 months of submission for standard applications.
Will biosimilars eventually replace biologics entirely?
Not entirely, but they’ll become the default choice for most patients. By 2030, analysts predict biosimilars could capture 40-50% of the market for biologic drugs. That’s up from 23% today. The goal isn’t to eliminate original biologics-it’s to give patients and providers affordable, effective options. Biologics will still be used when biosimilars aren’t available, or when a patient doesn’t respond to the alternative.








13 Comments
This is huge for patients like my mom who’ve been paying $3k/month for Humira. 💸❤️ She switched to the biosimilar and now pays $18. Same results. No more choosing between rent and medicine.
Let me guess-big pharma is gonna cry about ‘safety’ while they keep raking in billions. 🤡 The FDA’s finally waking up. Why are we still letting corporations hold patients hostage with price tags that make no sense? This isn’t medicine-it’s extortion.
I’ve seen too many people have ‘minor’ reactions that turned into full-blown flares. This is a slippery slope. The FDA is prioritizing speed over safety. And don’t get me started on pharmacists swapping drugs without consent. Patients deserve transparency, not convenience.
Analytical characterization via LC MS and bioassays sufficient for equivalence assessment when structural fidelity exceeds 99.999% and immunogenicity profiles match within 2SD of reference product variance. Clinical trials redundant for mAbs with well defined MoA
You think this is about patients? Nah. This is Big Pharma’s playbook. They let a few biosimilars in so they can bury the rest under patent thickets and lawsuits. Watch how fast the ‘interchangeable’ label gets revoked when the market gets crowded.
Bro this is the most exciting thing in healthcare since the vaccine rollout. I work in a clinic and we’ve saved over $200k in 6 months switching to biosimilars. Patients are thrilled. Docs are slowly coming around. Stop being scared of science.
It’s not just about cost... it’s about dignity. When someone has to choose between their treatment and their kid’s lunch money, that’s not a healthcare system. That’s a moral failure. The FDA’s doing what’s right-not what’s easy. I’m proud of this shift 🙏
If you’re still scared of biosimilars, you’re not being cautious-you’re being brainwashed by pharma ads. I’ve been on three different biosimilars. Same effect. Same side effects. Same life. And I’m alive because I didn’t let fear cost me my future.
In India we’ve had biosimilars for over a decade-cheaper, life-saving, and trusted. Why does the US act like this is some wild new experiment? It’s not. We’ve been doing this right for years. Time to catch up, not overthink.
The FDA’s guidance explicitly states that interchangeability requires separate regulatory review under 42 U.S.C. § 262(k)(4). The Commissioner’s statement is legally incoherent and potentially actionable. Mischaracterizing statutory requirements as ‘legislative terms’ is not scientific-it’s administrative overreach.
I’ve had patients ask me if biosimilars are ‘real’ drugs. I just show them the FDA’s data and their stories. Most are relieved. Some still worry. That’s okay. We just need to keep talking. No rush. No pressure. Just facts and patience.
omg i just switched to a biosimilar last month and i was so scared but my rheum doc said it was fine and now i pay like 20 bucks a month?? like i can actually buy groceries now?? 🥲❤️
The real story here isn’t the FDA’s new guidance-it’s the fact that 89% of U.S. hospitals now include biosimilars in their formularies. That’s institutional change. That’s systemic momentum. The science is solid. The data is clear. The only thing holding us back is the inertia of fear and outdated reimbursement models. We’re not just approving drugs-we’re redefining access. And it’s happening faster than anyone expected. The patients who’ve waited years for relief? They’re finally getting it. And no amount of patent litigation or state bureaucracy can undo what’s already in motion. This isn’t a policy tweak. It’s a revolution.